| OBSAH Úvod k dědičným onemocněním sítnice Dystrofie sítnice s časnou manifestací |
|
ÚVOD K DĚDIČNÝM ONEMOCNĚNÍM SÍTNICE Dědičná
onemocnění sítnice jsou velmi různorodá. Patří
mezi nejčastější příčiny slepoty u mladších
jedinců. Popsáno bylo více než 120 klinických
jednotek, z nichž nejčastější je retinitis
pigmentosa a nejzávažnější Leberova vrozená slepota.
V České republice nebyla tato onemocnění dosud
předmětem systematického výzkumu. Počet pacientů,
jejich diagnózy ani molekulárně genetické příčiny
nejsou tedy přesně známy. |
|
ANATOMIE SÍTNICE Sítnice
je uložena uvnitř oční koule a je zodpovědná za
vnímání světla. Na sítnici rozlišujeme vrstvu
vnitřní s fotoreceptory, nervovými vlákny a
pomocnými buňkami a vnější vrstvu pigmentového
epitelu. Zevně je pak uložena cévnatka.
|
|
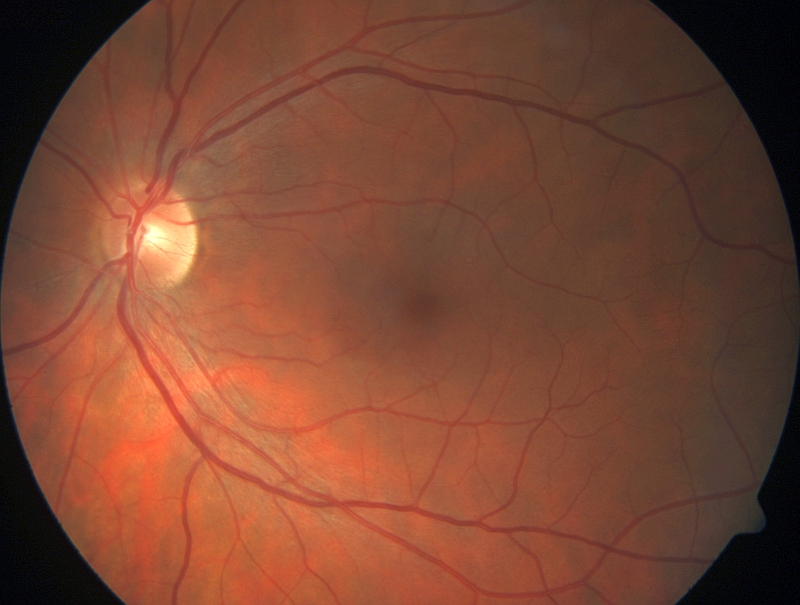
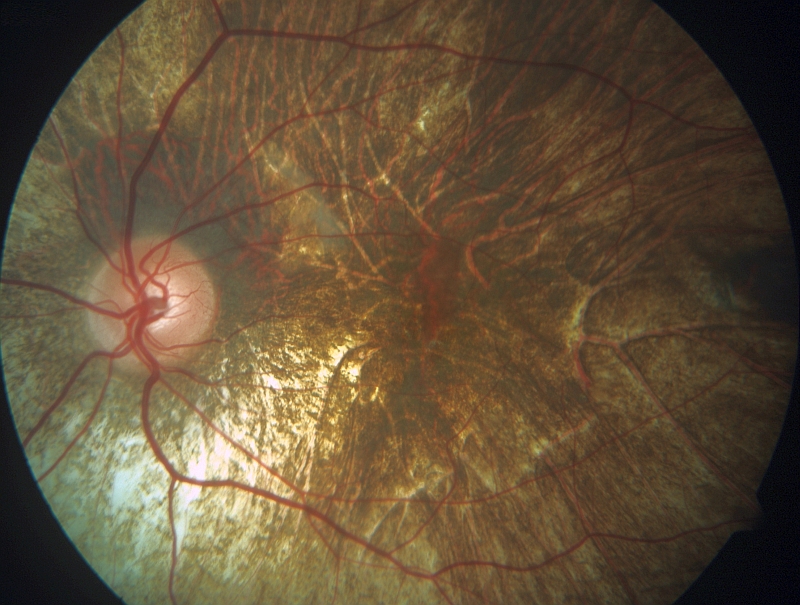
RETINITIS PIGMENTOSA Pod pojmem retinitis pigmentosa (RP), do češtiny někdy překládána také jako pigmentová retinopatie, je zahrnována celá řada různorodých onemocnění sítnice, jejichž společným znakem je primární postižení tyčinek. RP se vyskytuje buď jako samostatné onemocnění nebo jako součást řady syndromů, tj. kromě očního postižení se zjišťují í jiné příznaky, obvykle hluchota. Nejčastější nesyndromová RP postihuje přibližně 1 ze 4 000 obyvatel, v praxi to tedy znamená, že jenom v České republice trpí RP okolo 2 700 lidí. Onemocnění se typicky projevuje nejprve šeroslepostí, poté úbytkem zorného pole. V pozdějších fázích dochází také k odumírání čípků a tím ke snížení centrální zrakové ostrosti. U některých typů tohoto onemocnění je postižení tyčinek a čípků rovnoměrné. RP vykazuje při klinickém vyšetření velkou variabilitu, a to i mezi příslušníky jedné rodiny. Za typický znak je při vyšetření očního pozadí považován nález shluků pigmentu tvarem připomínající kostní buňky. Dále bývá přítomno zúžení tepen na sítnici, atrofie terče zrakového nervu, zákaly čočky a změny při elektroretinografickém (ERG) vyšetření. Onemocnění
může být přenášeno všemi známými typy
dědičnosti, tj. recesivně, dominantně nebo s vazbou
na pohlavní chromozóm. Vybraný odkaz na informace určené pacientům s retinitis pigmentosa.
|
|
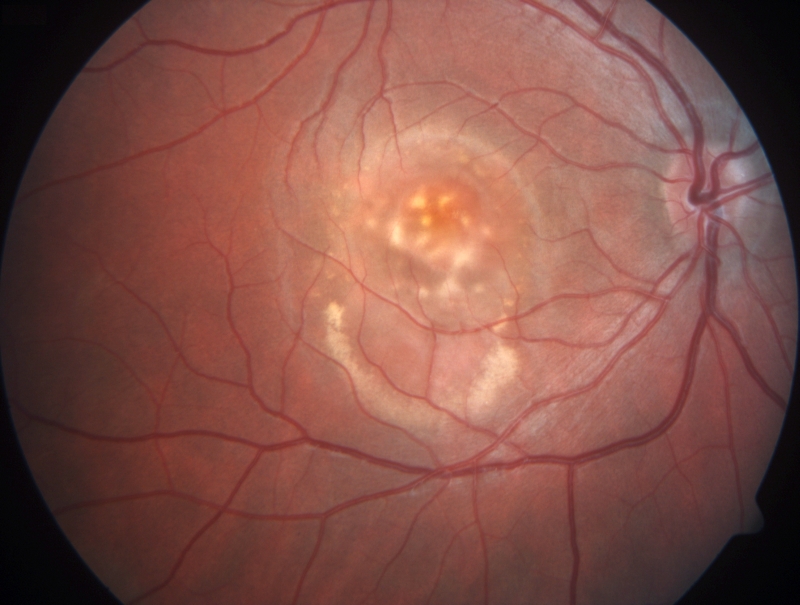
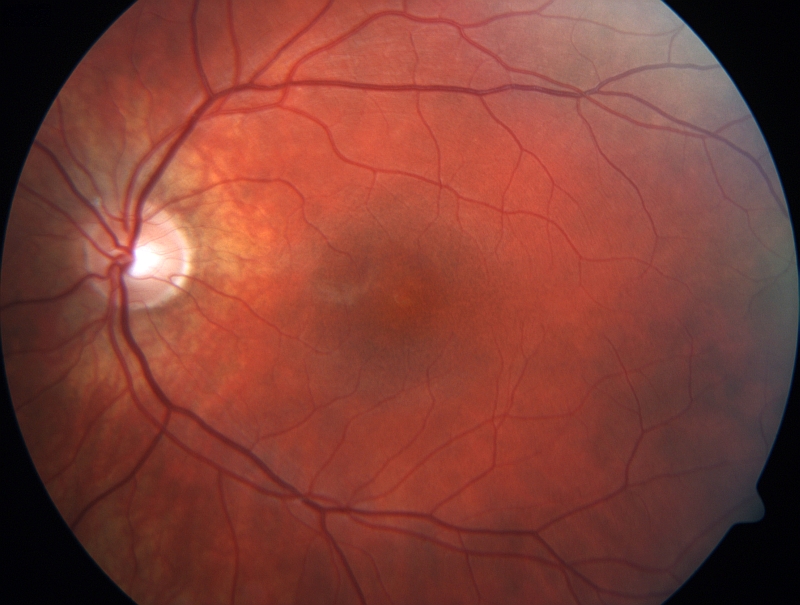
BESTOVA CHOROBA Bestova choroba (také známá jako viteliformní makulární dystrofie či Bestova viteliformní dystrofie makuly) je oboustranné onemocnění s výskytem jednoho nemocného na 10 000 obyvatel. Onemocnění má často rozdílný průběh na každém oku. Zpočátku je vzhled makuly normální, v další fázi zatím bez funkčních projevů jsou přítomny pouze jemné změny pigmentového epitelu sítnice. Bestova choroba se typicky projevuje mezi 10. až 30. rokem věku vznikem oranžovožlutého ložiska v makule, které svým vzhledem připomíná vaječný žloutek. V dalším vývoji může dojít k částečnému vstřebávání ložiska. V poslední fázi onemocnění, která je provázená často výraznějším poklesem zrakové ostrosti, může dojít ke vzniku jizvy nebo i k růstu nových cév pod nebo kolem jizvy. Ne všichni pacienti procházejí všemi fázemi onemocnění. Obtíže ve smyslu zkresleného a rozostřeného vidění a snížení centrální zrakové ostrosti mohou vznikat již v dětství, ale i o několik desetiletí později. V dlouhodobějším časovém horizontu dochází u většiny pacientů k poklesu zrakové ostrosti. Diagnóza je potvrzena ve většině případů zvýšenou autofluorescencí žlutých ložisek. Onemocnění se přenáší autozomálně dominantně a je podmíněno přítomností mutací v genu pro bestrofin 1, označovaném symbolem BEST1.
|
|
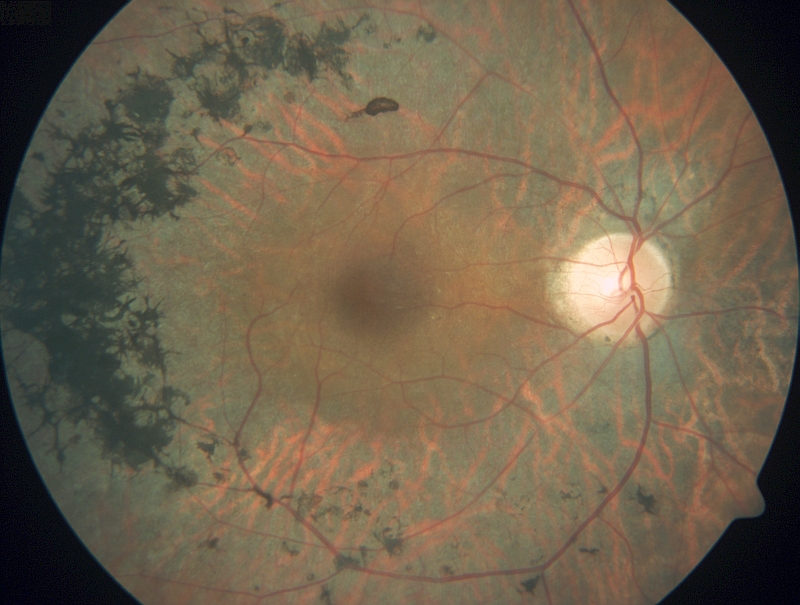
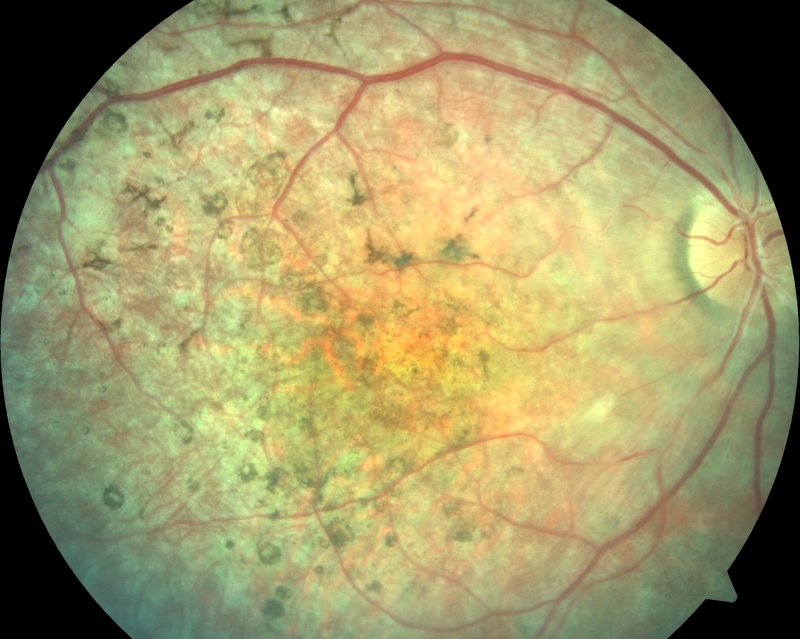
STARGARDTOVA CHOROBA Stargardtova choroba a fundus flavimaculatus jsou varianty dědičného onemocnění, které postihuje pigmentový epitel sítnice a fotoreceptory. Vyskytuje se přibližně u jednoho člověka na 10 000 obyvatel. Charakteristickým klinickým nálezem je přítomnost žlutých skvrn v makule spolu se ztenčením makuly a změnou její architektoniky. Stargardtova choroba se typicky projevuje snížením centrální zrakové ostrosti, které vzniká již v dětství nebo v časné dospělosti, známky onemocnění se ale mohou dostavit i později. Zpočátku může mít makula normální vzhled, následně nabývá vzhledu tepaného kovu a postupně vzniká jizva se žlutými skvrnami při okrajích nebo i po celém očním pozadí. Za nejspolehlivější metodu, jak rozpoznat Stargardtovu chorobu, je dnes považována autofluorescence očního pozadí. Optická koherenční tomografie, vyšetření zorného pole a fluorescenční angiografie se v diagnostice také používají a mohou poskytnout další užitečné informace. Onemocnění vykazuje v naprosté většině případů recesivní typ dědičnosti a je podmíněné mutacemi v genu ABCA4, který kóduje transportní mechanismy v tyčinkách a čípcích. Narušení transportu použitých částí fotoreceptorů způsobí jejich nahromadění v sítnici ve formě toxických sloučenin a následnému odumírání fotoreceptorů. Mutace v genu ABCA4 způsobují celou řadu degenerací s postižením makuly. Kromě Stargardtovy choroby jsou mutace v tomto genu zodpovědné rovněž za současné postižení tyčinek a čípků a mohou mít klinický obraz retinitis pigmentosa. Vybraný odkaz na informace určené pacientům se Stargardtovou chorobou a jejich rodinným příslušníkům.
|
|
CHOROIDEREMIE Choroideremie je onemocnění sítnice s dědičností vázanou na chromozom X a postihující 1 z 50 000 obyvatel. Chorobu způsobují mutace v genu CHM, jehož produkt reguluje transport vezikul do buněk a z buněk. Pacienti s choroideremií mají snížené nebo nulové množství tohoto enzymu, což vede k poruchám transportu zrakového pigmentu do fotoreceptorů, k poruchám migrace melanozomů v buňkách pigmentového epitelu sítnice a ke snížené fagocytóze zevních segmentů fotoreceptorů buňkami pigmentového epitelu sítnice. Následkem je degenerace choriokapilaris, pigmentového epitelu sítnice a fotoreceptorů s výrazným snížením zrakové ostrosti, postupně až úplnou ztátu zraku, která vzniká typicky v 5. až 7. dekádě. I přes intenzivní výzkum není dosud jasné, která část sítnice je u choroideremie primárně postižená. Brožura ke stažení (anglicky).
|
|
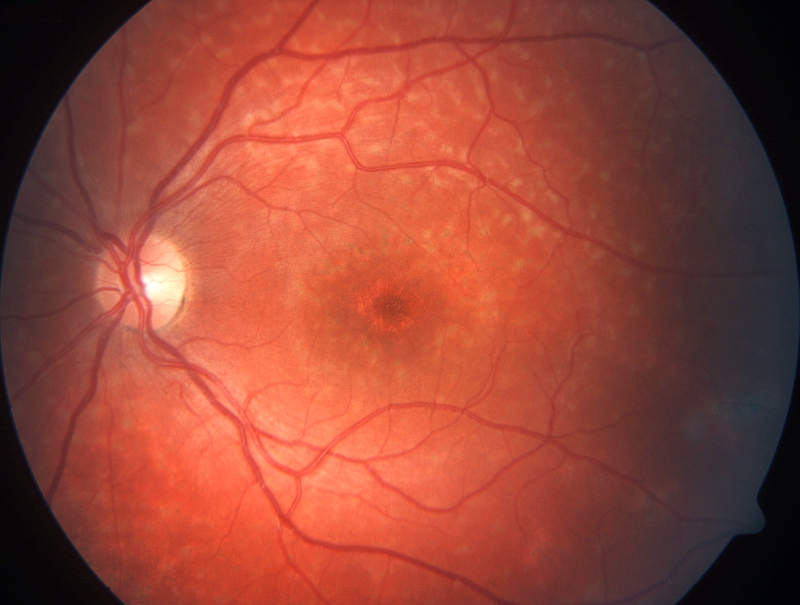
DYSTROFIE ČÍPKŮ Postižení čípků je provázeno poklesem až ztrátou centrální zrakové ostrosti, světloplachostí, poruchami barevného vidění a změnami při ERG vyšetření za světelných podmínek, celková elektrická odpověď sítnice pří ERG bývá zachovaná. Onemocnění se vyskytuje u jednoho člověka na 10-25 000 obyvatel. Většina postižených má první projevy onemocnění již v prvním desetiletí života. Oční pozadí může být téměř normální nebo s nepravidelnými změnami pigmentu v makule nebo s okrouhlou jizvou makuly. Optická koherenční tomografie prokazuje ztenčení centrální části sítnice s kompletním vymizením vrstev ve fovee, při fluorescenční angiografii nacházíme v centru sítnice obraz připomínající býčí oko. Dystrofie čípků vykazují všechny známé typy monogenní dědičnosti a na jejich vzniku se podílejí především mutace v genech RPGR, CNGA3, ABCA4.
|
|
ZÁVAŽNÉ DYSTROFIE SÍTNICE S ČASNOU MANIFESTACÍ Řada dystrofií sítnice se manifestuje v dětství. Pokud je onemocnění diagnostikováno na základě klinických známek do 6 měsíců života, označuje se jako Leberova kongenitální (vrozená) amauróza (slepota). V praxi je však tento termín často používán i pro popis klinického nálezu u pacientů s pozdějším nástupem onemocnění. Mezi hlavní projevy patří těžké postižení zraku, kmitavý pohyb očí, šilhání, chybění fixace, bloudivé pohyby očí, stlačování očí prsty. Závažné dystrofie sítnice s časnou manifestací se vyskytují u jednoho člověka na 80 000 obyvatel. Dosud bylo zjištěno nejméně než 25 příčinných genů. Přibližně u 5 % pacientů jsou přítomny molekulárně genetické změny v genu RPE65, u kterého probíhaly od roku 2007 úspěšné klinické zkoušky s genovou terapií, které vyústily v roce 2017 ve schválení první genové terapie pro klinickou oftalmologickou praxi.
|
|
JAKÉ JSOU MOŽNOSTI LÉČBY SÍTNICE? Možnosti
léčby dědičných onemocnění sítnice jsou zatím
velmi omezené. Ke zpomalení progrese onemocnění se v
některých zemích pacientům s RP doporučovalo
podávání vysokých dávek vitamínu A, další
klinické studie zaměřené na suplementaci jinými
látkami, např. luteinem, či ochranou sítnice před
oxidativním poškozením probíhají. Znalost
molekulárně genetické příčiny zlepšuje klinické
poradenství pacientům a jejich rodinám a umožňuje
postiženým jedincům s vysokým rizikem přenosu
onemocnění na potomstvo podstoupit preimplantační
diagnostiku (více o preimplantační diagnostice na
tomto odkazu) a tak zabránit dalšímu přenosu
onemocnění. Na základě příznivých výsledků z
klinických studií u pacientů s mutacemi v RPE65 byla
v roce 2017 v Americe a v roce 2018 v Evropě schválena
první genové terapie (více na tomto
odkazu). Nadějné jsou také studie
zaměřující se na výzkum kmenových buněk. |
|
VÝZKUM ROHOVKOVÝCH DYSTROFIÍ Rohovkové
dystrofie je skupina více než 30 onemocnění
projevující se různými zákaly rohovky, která se
dědí nejčastěji autozomálně dominantně. Monogenně
dědičná onemocnění rohovky jsou vzácná, tj.
zjišťována u méně než 5 na 10 000 obyvatel. Do
této skupiny chorob řadíme rohovkové dystrofie a
různá vývojová onemocnění rohovky. V české populaci jsme zatím úspěšně klinicky charakterizovali a příčinné mutace zjistili u pacientů s granulární dystrofií rohovky, Reisovou-Bucklersovou dystrofií, dystrofií rohovky Thiela-Behnkeho, mřížkovou dystrofií rohovky, makulární dystrofií rohovky, Harboyanova syndromu (kongenitální hereditární dystrofie rohovky asociovaná s percepční hluchotou) a se zadní polymorfní dystrofií rohovky. Vývojové anomálie rohovky zahrnují onemocnění megalokornea a mikrokornea. Megalokornea vzniká na podkladě mutací v genu CHRDL1, který se nalézá na pohlavním chromozómu. Ve velmi vzácných případech je megalokornea asociována s mentální retardací, tzv. Neuhauserův syndrom. Genetická příčina tohoto syndromu zůstává neobjasněna, onemocnění se dědí pravděpodobně autosomálně recesivně. Nesyndromový keratokonus je dnes považován za komplexní onemocnění, na jehož vzniku se podílí i řada genetických faktorů. Přibližně v 10 % případů je postiženo více rodinných příslušníků a v některých rodinách se předpokládá, že přenos onemocnění vykazuje Mendelovskou dědičnost, nicméně, dosud nebyl zjištěn žádný gen, který by byl jednoznačně prokázán jako příčinný. Zadní polymorfní dystrofie rohovky vzniká na podkladě mutací ve třech genech ZEB1, OVOL2 a GRHL2, na objevech dvou z těchto genů se skupina doc. Liškové ve spolupráci s britským týmem zásadně podílela. Například v genu OVOL2 se přičinné mutace nacházejí v nekódující sekvenci, trvalo pátrání po tomto genu celých 20 let. Zadní polymorfní dystrofie rohovky je vzácné onemocnění, nicméně na našem územi je jeho prevalence největší na světě díky tzv. zakladatelskému efektu. Většina postižených jedinců sdílí stejnou mutaci, kterou získali od společného předka a jejich rodiny pocházejí z okoli Klatov. |
|
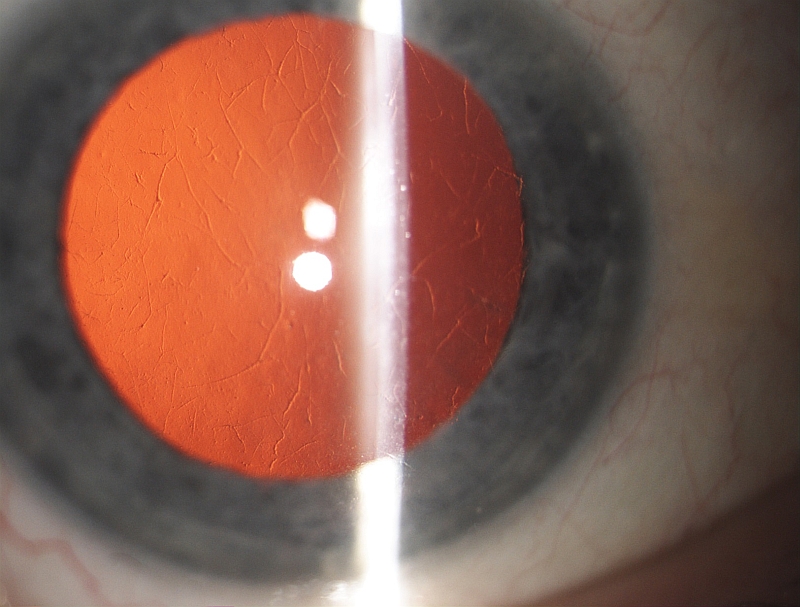
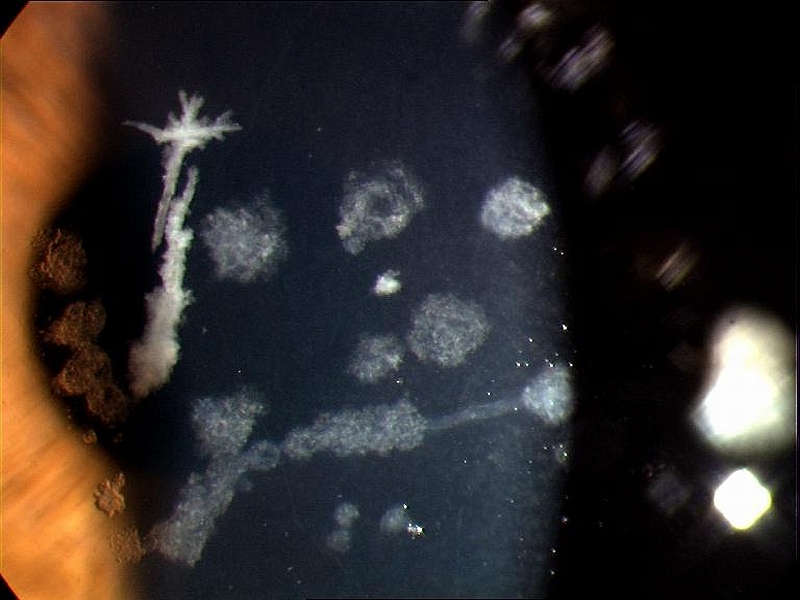
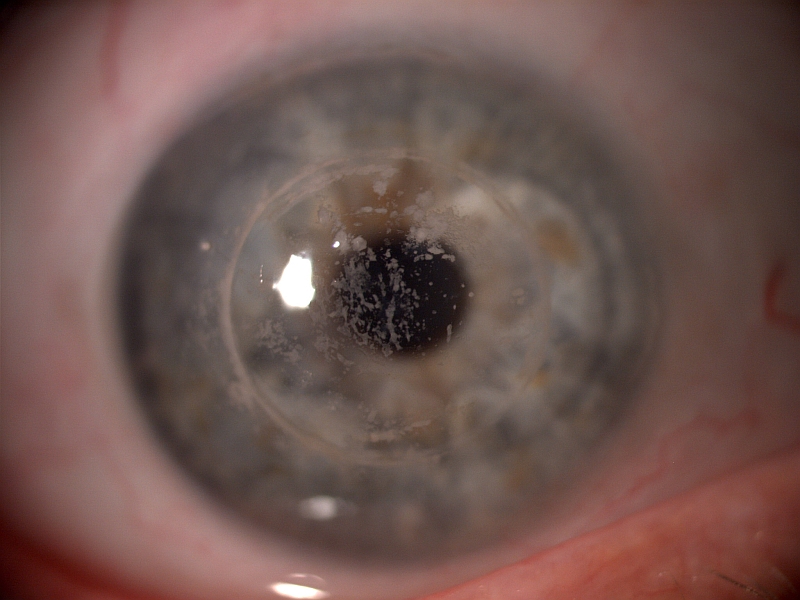
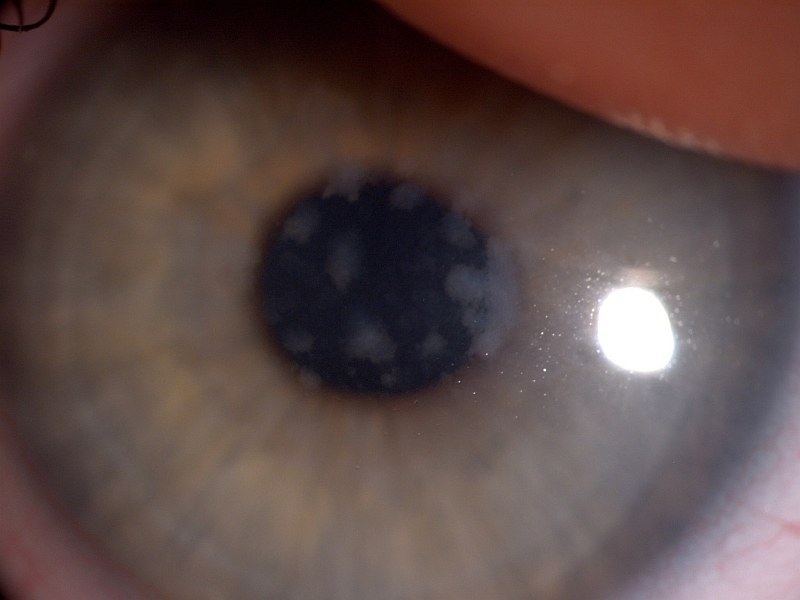
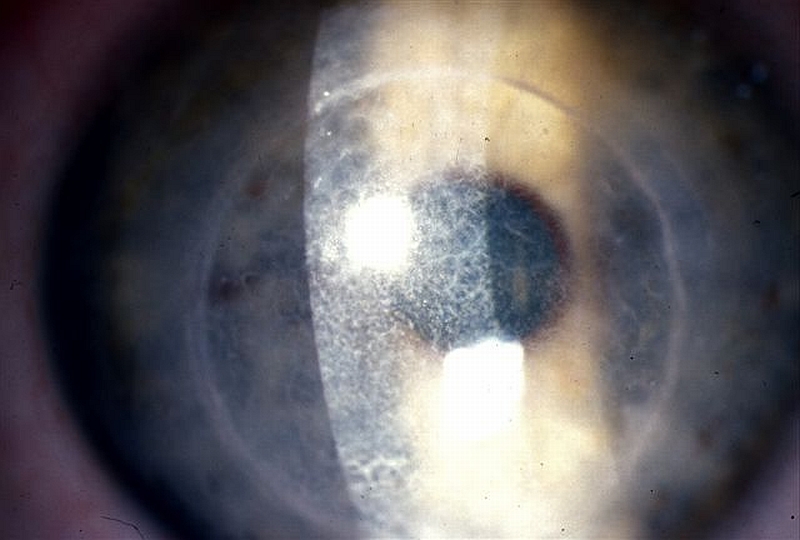
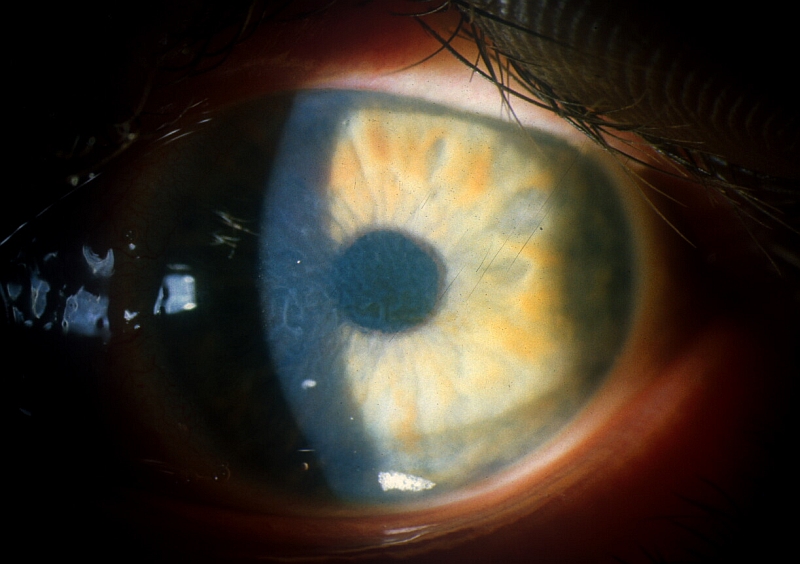
PŘÍKLADY NÁLEZŮ NA ROHOVKÁCH
|
|
Máte-li zájem se našeho výzkumu zúčastnit, vyplňte, prosím, kontaktní formulář. |
|